 2016, Vol. 36
2016, Vol. 36



215123 苏州大学医学部放射医学与防护学院(孙阳、涂彧)
School of Radiation Medicine and Protection, Medical School of Soochow University, Suzhou 215123, China (Sun Y, Tu Y)
放射性脑损伤(RBI)作为颅脑部肿瘤患者进行放射治疗后的常见并发症,严重影响患者的治疗效果及生活质量。研究发现,炎症反应在RBI的发生发展中扮演了重要的角色,是脑组织受到电离辐射后的急性损伤和晚期继发性损伤的主要因素[1]。环氧合酶-2(Cox-2)是一种可诱导酶,其既受各种生长因子及炎性介质的调控,也介导合成多种前列腺素类物质,进而生成多种炎性因子,以瀑布式级联反应参与机体各生理、病理过程,临床上针对此靶点已有多种成熟的抗炎药物[2]。本研究拟通过基因、蛋白及功能定位等来探讨Cox-2在RBI中的动态变化规律,希冀为临床上使用选择性Cox-2抑制剂治疗RBI提供一定的思路及依据。
1. 实验动物饲养与分组:选用SPF级健康雄性SD大鼠112只,8周龄,体重(290±12)g,分笼饲养于苏州大学实验动物中心。SD大鼠购于苏州大学实验动物中心,许可证编号:2013001809303。采用随机数字表 法,将动物分为健康对照组(56只,麻醉不照射)和照射组(56只,麻醉同时予全脑单次20 Gy照射)。
2. 主要实验仪器及试剂:NanoDrop 分光光度计(美国Thermo公司);梯度PCR仪(德国BioMetra公司);实时荧光定量PCR仪:ViiA7(美国LifeTechnologies公司);光学显微镜(德国莱卡公司);Primus M医用直线加速器(德国Siemens公司);Synergy2多功能酶标仪(美国BioTek公司)。
TRIzol试剂(美国Invitrogen公司);RT-PCR试剂盒(日本TaKaRa公司);Taqman探针、引物及反应预混液(瑞士Roche公司);Cox-2兔多克隆抗体(英国Abcam公司);BCA标准蛋白试剂盒(美国Thermo Scientific公司);Western blot试剂盒、电化学发光(ECL)显色试剂盒、β-肌动蛋白单抗、兔鼠二抗等购于江苏碧云天公司;其他一些常用试剂购于国药集团化学试剂有限公司。
3. 照射方法:将大鼠给予10%水合氯醛,剂量0.3 ml/100 g腹腔麻醉后俯卧置于治疗床上,模拟机确定照射野后,以挡铅块保护照射野外组织。采用医用直线加速器,给予5 MeV电子线行垂直照射,取脑中平面作为等中心平面,采用单次全脑大剂量照射,总吸收剂量为20 Gy,吸收剂量率为200 cGy/min,源皮距为100 cm。于照射后3、12 h,1、3、7、15及30 d取大鼠全脑组织。
4. 大鼠脑组织病理学观察:大鼠于预设观测时间点处死后,取脑组织切片行苏木素-伊红(HE)染色,观察受照射后大鼠炎症反应过程中的组织学病理变化。
5. 实时荧光定量反转录聚合酶链反应(QRT-PCR)检测脑组织中Cox-2 mRNA表 达水平:于观测时间点处死大鼠后,取其海马体及周围脑组织,剪碎后在液氮冷冻的同时于研钵中研磨成干粉状,加入1 ml TRIzol裂解细胞以提取脑组织中的总RNA。NanoDrop 分光光度计检测样品A260/A280的比值及浓度,用灭菌后的DEPC-H2O调节总RNA浓度至0.5~1.0 μg/μl。梯度PCR仪中反转录合成cDNA,合成条件为37℃ 15 min,85℃ 5 s,4℃ 1 h。利用实时荧光定量ViiA7 PCR仪行PCR反应,按mRNA荧光定量检测试剂盒配制反应液,反应条件为预变性:95℃ 30 s,变性:95℃ 5 s,退火/延伸:57℃ 30 s,累计40个循环。引物序列见表 1,GAPDH为内参。
| 表 1 Cox-2基因引物序列 Table 1 The primer sequences of Cox-2 gene |

6. Western blot法检测大鼠脑组织中Cox-2蛋白表 达量:于照射后各个观察时间点处死大鼠,取全脑组织,用适量裂解液提取脑组织的总蛋白,BCA法测定蛋白浓度。10% SDS-PAGE凝胶电泳分离蛋白样本。电泳条件为浓缩胶电压80 V,分离胶电压120 V。转膜条件为聚偏氟乙烯(PVDF)湿转法,恒定电流300 mA,2.5 h。5% BSA室温封闭2 h。蛋白一抗4℃孵育16 h,TBST缓冲盐溶液洗膜15 min × 4次,二抗室温孵育100 min,TBST洗膜10 min × 4次。ECL化学显色试剂盒显影,化学发光凝胶成像分析仪曝光成像。本实验以β-肌动蛋白为内参对照。
7. 免疫组织化学技术法检测大鼠海马区中Cox-2蛋白表 达情况:于照射后各预设时间点处死大鼠,断头取全脑置于10%的甲醛固定液中固定24 h,石蜡包埋、切片、脱蜡水化、封片,用光学显微镜观察及采集图 像。
8. 统计学处理:实验数据以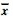 ±s表 示。采用SPSS 19.0 软件进行数据分析,两组间比较采用独立样本t检验。P<0.05为差异有统计学意义。
±s表 示。采用SPSS 19.0 软件进行数据分析,两组间比较采用独立样本t检验。P<0.05为差异有统计学意义。
1. 大鼠RBI模型脑组织病理学变化特征:单次大剂量照射后大鼠脑组织的HE染色切片结果如图 1所示。健康对照组脑组织未见明显的病理变化,各组织结构完整,层次清晰,细胞形态完整,血管形状完整。RBI模型中可见照射后3 h即有少量细胞出现胞质水肿、核碎裂、固缩等现象。照射后12 h,1、3 d可见水肿细胞数目进一步增多,胞质空泡化和脂肪变性更加明显,毛细血管间隙增宽,部分血管内有血栓形成。照射后7 d病变细胞数达到最高,部分小血管周围脑组织结构变得紊乱。照射后15 d可见部分细胞坏死,细胞核破碎或形状不规则,血管栓塞现象仍存在,部分脑组织因血管栓塞导致缺血梗死。照射后30 d可见栓塞血管多已机化再通,水肿细胞已消失,坏死区域较前稍有增多。

|
图 1 单次大剂量照射后大鼠脑组织切片 HE染色 ×100 A. 健康对照组;B-H. 依次为照射组照后3、12 h,1、3、7、15、30 d Figure 1 Image of rat brain tissue after a single high dose irradiation HE staining ×100 A. Healthy control group; B-H. Irradiated group at 3 and 12 h, 1, 3, 7, 15 and 30 d after irradiation, respectively |
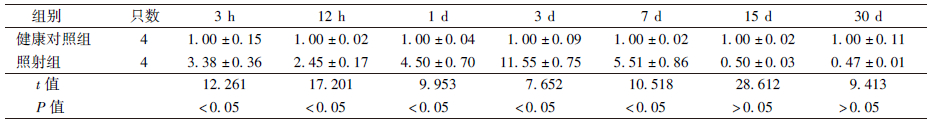
2. 大鼠RBI模型中Cox-2 mRNA的表 达变化:单次大剂量照射后不同时间大鼠脑组织中Cox-2 mRNA表 达的变化结果列于表 2。由表 2可知,大鼠脑组织中Cox-2 mRNA的表 达在受照射后3 h开始增加,照射后12 h表 达量稍有下降,照射后第3天表 达达到高峰,随后表 达量逐渐减少,照射后第15、30天表 达量稍低于正常水平。与健康对照组相比,照射后3、12 h,1、3、7 d的表 达量均差异有统计学意义(t=7.652~17.201,P<0.05)。
| 表 2 单次大剂量照射后不同时间大鼠脑组织中Cox-2 mRNA表 达的变化(±s)
Table 2 The expression of Cox-2 mRNA in rat brain tissue at different time after a single high dose irradiation(±s) |
3. Western blot检测Cox-2蛋白表 达水平:单次大剂量照射后大鼠脑组织中Cox-2蛋白表 达变化的Western blot检测结果如图 2所示。Cox-2在健康对照组的大鼠脑组织中表 达量极低,但在受到辐射处理后表 达量随着时间的推移逐渐增多,在照射后第3天Cox-2蛋白的表 达量达到高峰,随后表 达量开始减少,至照射后30 d稍高于正常水平。

|
图 2 单次大剂量照射后大鼠脑组织中Cox-2蛋白表 达变化结果 Figure 2 Expression of Cox-2 protein in rat brain tissue after a single high dose irradiation |
4. 免疫组织化学检测Cox-2蛋白表 达水平结果:单次大剂量照射后大鼠脑组织中Cox-2蛋白表 达变化的免疫组织化学检测结果如图 3所示。选取大鼠脑组织海马区为主要观测区域,图 中呈棕褐色黄染的细胞即为Cox-2蛋白表 达阳性的细胞,且蛋白主要分布于细胞质中。结果可见健康对照组几乎无Cox-2蛋白表 达,照射后3 h既有少量Cox-2蛋白表 达,随着时间推移,阳性表 达量逐步增多,至3 d Cox-2表 达达到高峰,随后开始减少。15、30 d仅可见少量阳性表 达和组织结构改变。

|
图 3 单次大剂量照射后大鼠脑组织中Cox-2蛋白表 达变化的免疫组化检测结果 ×100 A. 健康对照组;B-H依次为照射后3、12 h,1、3、7、15、30 d组 Figure 3 The immunohistochemical image of Cox-2 protein in rat brain tissue after a single high dose irradiation ×100 A. Healthy control group; B-H. Irradiated group at 3 and 12 h, 1, 3, 7, 15 and 30 d after irradiation, respectively |
RBI是一个动态进展的病理过程,包括射线引起的胶质细胞及神经元细胞损伤和免疫炎性反应所致的血管闭塞性改变,组织水肿,功能障碍等[3]。本研究的RBI模型HE染色结果中,急性期的脑损伤以细胞水肿、微血管通透性增加、血管内膜增生、管腔狭窄和微血栓形成引起局部脑组织缺血缺氧等病理改变为主,而在中晚期则以脑胶质细胞的坏死、凋亡、脱髓鞘、组织结构紊乱等为主,与文献报道基本一致。
前期预实验的过程中以10、15、20和25 Gy探索实验条件,结果显示10和15 Gy的照射剂量对大鼠的脑损伤不明显,而25 Gy的剂量则过高,大鼠的死亡率达到了近40%,而20 Gy的照射剂量既可保证成功构建大鼠放射性脑损伤模型,且不会导致大鼠因照射而死亡。因此,照射组在麻醉同时予全脑单次20 Gy照射。
本研究结果显示,大鼠正常脑组织在接受照射后3 h,即可观测到Cox-2 mRNA及蛋白质的表 达量增加,至照射后第3天达到峰值,随后逐渐减少,至照射后30 d稍高于正常水平。其原因是射线可以诱导Cox-2的表 达并参与了RBI的发生发展。 Cox-2在正常情况下不表 达或表 达量极低,但在受到一定的外因刺激后,其表 达量可以显著增高,参与诸多生理病理反应[4, 5, 6]。在RBI进程中,射线诱导了Cox-2的表 达,上调的Cox-2进一步介导前列腺素E2(PGE2)的生成,PGE2具有趋化白细胞富集,促进局部血管扩张,引起毛细血管通透性增加,诱发红、肿、热、痛及功能障碍的作用[7]。PGE2还可以促进诸多炎性因子如肿瘤坏死因子-α(TNF-α)、白细胞介素-1β(IL-1β)、细胞间黏附因子-1(ICAM-1)等的表 达,这些炎性因子可以损伤血管内皮细胞,引起局部血管坏死导致出血,诱导T、B淋巴细胞增殖活化,参与免疫炎性应答,促使白细胞黏附于血管内皮细胞,使血管内皮细胞间隙增加,组织液渗出,引起细胞周围间隙水肿,微环境改变等病理表 现[8]。在血管损伤方面,Cox-2可以介导血栓素A2(TXA2)的生成,TXA2是一种强血管收缩剂,可以激活血小板,使其大量聚集形成血栓,黏附在血管壁上,使管腔狭窄、管壁增厚,影响脑血管的舒缩,最终导致栓塞血管周围的脑组织缺血、缺氧,形成梗死灶[9]。缺氧又可以使脑组织能量代谢发生障碍,抑制细胞膜上的钠/钾ATP酶活性,影响细胞膜内外离子转运,形成细胞内高渗状态,大量水分内流入胞内,引发细胞毒性水肿。此外,Cox-2还可以通过其氧化酶的活性促进活性氧簇(ROS)的生成,增加的ROS可以与脑组织中的不饱和脂肪酸反应,引起脂质过氧化,进而损伤细胞。Covey等[10]也发现降低生物体内的Cox-2表 达量可以有效减轻ROS导致的继发性氧化应激性脑损伤。
在肿瘤研究领域,有研究发现,Cox-2表 达量的高低与多种肿瘤的分化程度、血管生成密切相关,甚至可能参与肿瘤的转移及耐药性形成[11, 12]。在肿瘤血管生成方面,有研究发现,上调Cox-2可以影响血管内皮细胞生长因子(VEGF)的表 达[13]。VEGF是有效的促血管生长因子,可以活化内皮细胞使其增殖重排成条索状,并刺激成纤维细胞分泌细胞外基质,形成新生血管,但这些新生的血管大多排列疏松,渗透性较强,易引起组织液的渗出,导致局部水肿。针对此信号通路来抑制肿瘤新生血管的形成,切断其营养供给途径,目前已成为一种治疗肿瘤的新思路[14]。但是,Cox-2具体是如何影响VEGF的表 达,具体通过何种通路实现,目前还未研究清楚。在关于肿瘤治疗的方面,一些临床研究发现,在放疗的同时使用选择性Cox-2抑制剂塞来昔布,降低肿瘤细胞内的Cox-2表 达水平有助于提高放疗效果[15, 16]。Zhang等[17]发现应用低剂量的塞来昔布抑制Cox-2的表 达,可以诱导肿瘤细胞在分裂过程中停滞于G2/M期和细胞凋亡,达到放射增敏的作用。有研究显示抑制Cox-2的表 达还可以抑制细胞的增殖,诱导肿瘤细胞凋亡,降低正常细胞的癌变率[18, 19]。这些研究表 明在应用选择性Cox-2抑制剂治疗放射性脑损伤的同时,不但可以有效降低脑组织细胞内的Cox-2表 达水平,同时亦可以通过降低颅脑部肿瘤细胞内的Cox-2含量,从而提高肿瘤的放疗效果,达到双赢的作用。
在大鼠RBI模型中,本研究了解Cox-2 mRNA及蛋白的变化规律,发现了Cox-2在炎症反应、血管损伤与修复等事件中都发挥了重要作用。因此,可以认为Cox-2有可能成为RBI的治疗靶点,早期、及时、针对性的使用一些Cox-2抑制剂,将可以有效的减轻RBI过程中的炎症损伤、血管病变,改善受损部位血供,提高肿瘤细胞对射线的敏感性,达到减轻患者的痛苦,提高治疗效果的目的。
利益冲突 本研究未接受其他机构提供的不当利益,与其他单位没有引起利益的冲突关系
作者贡献声明 熊耀祖参与研究的酝酿和设计实验,具体实施,采集数据,分析处理,论文撰写与修改;孙阳参与研究的数据分析;涂彧参与研究的酝酿和设计实验;徐晓婷参与研究的酝酿和设计实验,数据分析,提供文章修改意见等
| [1] | Harting MT, Jimenez F, Adams SD, et al. Acute, regional inflammatory response after traumatic brain injury: implications for cellular therapy[J]. Surgery, 2008, 144(5): 803-813. DOI: 10.1016/j.surg.2008.05.017. |
| [2] | Kalinski P. Regulation of immune responses by prostaglandin E2[J]. J Immunol, 2012, 188(1): 21-28. DOI: 10.4049/jimmunol.1101029. |
| [3] | Kim JH, Brown SL, Jenrow KA, et al. Mechanisms of radiation-induced brain toxicity and implications for future clinical trials[J]. Neurooncol, 2008, 87(3):279-286. DOI: 10.1007/s11060-008-9520-x. |
| [4] | Khan Z, Khan N, Tiwari RP, et al. Biology of Cox-2: an application in cancer therapeutics[J]. Curr Drug Target, 2011, 12(7): 1082-1093. DOI: 10.2174/138945011795677764. |
| [5] | Sellers RS, Radi ZA, Khan NK.Pathophysiology of cyclooxygenases in cardiovascular homeostasis[J]. Vet Pathol, 2012, 47(4): 601-613. DOI: 10.1177/0300985810364389. |
| [6] | Murono S, Inoue H, Tanabe T, et al. Induction of cyclooxygenase-2 by epstein-barr virus latent membrane protine 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells[J]. Proc Natl Acad Sci USA, 2001, 98(12): 6905-6910. DOI: 10.1073/pnas.121016998. |
| [7] | Ji K, Tsirka SE. Inflammation modulates expression of laminin in the central nervous system following if chemic injury[J]. J Neuroinflamm, 2012, 9: 159. DOI: 10.1186/1742-2094-9-159. |
| [8] | Ramanan S, Kooshki M, Zhao W, et al. PPAR α ligands inhibit radiation-induced microglial inflammatory response by negatively regulating NK-κB and AP-1 pathways[J].Free Radic Biol Med, 2008, 45(12): 1695-1704. DOI: 10.1016/j.freeradbiomed.2008.09.002. |
| [9] | Lucas J, Mack WJ. Effects of ionizing radiation on cerebral vasculature: a review[J]. World Neurosurg, 2014, 81(3-4): 490-491. DOI: 10.1016/j.wneu.2014.01.006. |
| [10] | Covey MV, Loporchio D, Buono KD, et al. Opposite effect of inflammation on subventricular zone versus hippocampal precursors in brain injury[J]. Ann Neurol, 2011, 70(4): 616-626. DOI: 10.1002/ana.22473. |
| [11] | Omura N, Griffith M, Vincent A, et al. Cyclooxygenase-deficient pancreatic cancer cells use exogenous sources of prostaglandins[J]. Mol Cancer Res, 2010, 8(6): 821-832. DOI: 10.1158/1541-7786. |
| [12] | Hill R, Li Y, Tran LM, et al. Cell intrinsic role of cox-2 in pancreatic cancer development[J]. Mol Cancer Ther, 2012, 11(10): 2127-2137. DOI: 10.1158/1535-7163. |
| [13] | Toomey DP, Murphy JF, Conlon KC, et al. Cox-2, VEGF and tumour angiogenesis[J]. Surgeon, 2009, 7(3): 174-180. DOI: 10.1016/S1479-666X(09)80042-5. |
| [14] | Nieves BJ, D'Amore PA, Bryan BA. The function of vascular endothelial growth factor[J]. Biofactors, 2009, 35(4): 332-337. DOI: 10.1002/biof.46. |
| [15] | Shin YK, Park JS, Kim HS, et al. Radiosensitivity enhancement by celecoxib, a cyclooxygenase (Cox)-2 selective inhibitor, via Cox-2-dependent cell cycle regulation on human cancer cells expressing differential Cox-2 levels[J]. Cancer Res, 2005, 65(20): 9501-9509. DOI: 10.1158/0008-5472. |
| [16] | Mutter R, Lu B, Carbone DP, et al. A phase Ⅱ study of celecoxib in combination with paclitaxel, carboplatin, and radiotherapy for patients with inoperable stage Ⅲ a/b non-small cell lung cancer[J]. Clin Cancer Res, 2009, 15(6): 2158-2165. DOI: 10.1158/1078-0432. |
| [17] | Zhang SX, Qiu QH, Chen WB, et al. Celecoxib enhances radiosensitivity via induction of G2/M phase arrest and apoptosis in nasopharyngeal carcinoma[J]. Cell Physiol Biochem, 2014, 33(5): 1484-1497. DOI: 10.1159/000358713. |
| [18] | Liu B, Wen JK, Li BH, et al. Celecoxib and acetylbritannilactone interact synergistically to suppress breast cancer cell growth via cox-2-dependent and -independent mechanisms[J]. Cell Death Dis, 2011, 2: e185. DOI: 10.1038/cddis.2011.64. |
| [19] | Sinicrope FA, Gill S. Role of cyclooxygenase-2 in colorectal cancer[J]. Cancer Metastasis Rev, 2004, 23(1-2): 63-75. DOI: 10.1023/A:1025863029529. |