 2015, Vol. 35
2015, Vol. 35



2. 苏州大学医学部放射医学与防护学院
细胞因子,尤其是炎性细胞因子在放射性脑损伤的发生发展过程中的作用,越来越受到关注[1]。进一步阐明其在放射性脑损伤发生过程中的作用,可以为防治放射性脑损伤的发生提供新的思路与方法。核转录因子-κB(NF-κB)是一种具有多向转录调节功能的核蛋白因子,广泛存在于机体多种细胞中,可调控免疫炎症反应、病毒相关、原癌基因等相关基因转录表达,从而调节免疫、炎症、增生、凋亡等重要生命活动[2, 3, 4],为调节炎性因子基因表达的关键转录因子之一[5]。本研究通过基因、蛋白、功能定位3个方面来观察NF-κB在放射性脑损伤发生过程中的动态变化,进一步探讨放射性脑损伤发生的机制,为NF-κB抑制剂在防治放射性脑损伤发生的应用提供理论依据。
1. 实验动物:Sprague-Dawley大鼠(SD大鼠)购于上海斯莱克斯实验动物有限责任公司,许可证编号SCXK(沪)2012-0002。健康雄性SD大鼠82只,周龄8周,体重(334±18)g,于安静、温暖(22℃)、光线柔和的环境下分笼饲养48 h,自由进食。
2. 主要试剂与仪器:TRIzol试剂购于美国Invitrogen公司;RT-PCR试剂盒购于日本TaKaRa公司;引物购于瑞士Roche公司;NF-κB单抗购自英国Abcam公司;BCA标准蛋白试剂盒购于美国Thermo Scientific公司;Western blot试剂盒、电化学发光(ECL)化学显色试剂盒、β-肌动蛋白单抗、山羊抗小鼠IgG购于江苏碧云天公司;NF-κB ELISA检测试剂盒购于武汉优尔生公司。NanoDrop分光光度计(美国Thermo公司);ABI 7500定量PCR仪(美国ABI公司);GL2200 Pro化学发光凝胶成像分析仪(美国Kodak公司);光学显微镜(德国莱卡公司);Primus M医用直线加速器(德国Siemens公司);Synergy2多功能酶标仪(美国BioTek公司)。
3. 动物分组及观察时间点的确立:利用随机数字表法,将动物分为健康对照组(32只,给予相同的麻醉但不照射),照射组(50只,单次全脑照射20 Gy),并于照射后1、3、7、14、28 d处死,断头取脑后进行相应的观察、检测。
4. 照射方法:大鼠用10%的水合氯醛经腹腔麻醉,剂量为0.35 ml/100 g体重,俯卧位,固定在自制的固定板上。照射前于模拟机下定位,确定照射部位(大体部位为上界:双眼后眦连线,下界:双耳后连线,两侧露空)及面积(设野2 cm×3 cm),照射野以外的部位用铅块遮挡。参照文献[6]建立大鼠急性放射性脑损伤(RBI)模型,采用医用直线加速器,应用5 MeV的电子线做全脑前后对穿垂直照射,等中心平面为脑中平面,采用单次全脑大剂量照射,总吸收剂量为20 Gy,吸收剂量率为3 Gy/min,源皮距100 cm。照射前校正剂量,以确保大鼠脑部总剂量的吸收。
5. 实时荧光定量反转录聚合酶链反应(QRT-PCR)检测脑组织中NF-κB mRNA表达水平:于照射后各个观察的时间点处死动物,每组每个时间点3只。断头取出脑组织,用研钵在液氮冷冻的情况下将剪碎的脑组织研磨成粉末状,加入1 ml的TRIzol提取脑组织中的总RNA,用NanoDrop分光光度计检测A260/A280的比值(比值在1.8~2.0之间表明样本处理可用)及浓度,用焦碳酸二乙酯(DEPC)-H2O调节总RNA浓度至0.5~1.0 μg/μl。按照RT-PCR试剂盒说明进行反转录合成cDNA,37℃反应15 min,85℃反应5 s,4℃保温1 h。利用ABI 7500定量PCR仪进行PCR反应,按照mRNA荧光定量检测试剂盒配制反应试剂,反应按95℃预变性10 min,95℃变性15 s,57℃退火30 s,72℃延伸30 s,累计40个循环。引物序列如表1。GAPDH为内参。
| 表1 引物序列 |

6. Western blot法检测脑组织中NF-κB蛋白表达水平:于照射后各个观察的时间点处死动物。断头取出脑组织,用适量裂解液提取脑组织的总蛋白,标准蛋白法测定蛋白浓度。10% SDS-PAGE凝胶电泳分离蛋白样本,浓缩胶电压80 V,分离胶电压120 V。聚偏氟乙烯(PVDF)转膜,湿转,恒流300 mA,120 min。5% BSA室温封闭90 min。蛋白一抗4℃孵育过夜,TBST缓冲盐溶液洗膜,二抗室温孵育1 h,TBST洗膜。ECL显色试剂盒显影,化学发光凝胶成像分析仪曝光成像,并用AlphaView SA 软件进行灰度值比较分析。
7. ELISA法检测脑组织中NF-κB蛋白表达水平:于照射后各个观察的时间点处死动物,每组每个时间点3只。取适量的脑组织,用预冷的1×PBS进行充分研磨,此步骤需在冰上进行,4℃,10 000 r/min离心15 min,离心半径10 cm,取上清。标准蛋白法测定上清蛋白含量,用1×PBS稀释上清至蛋白终浓度为1 500 μg/ml。样品中的NF-κB的检测按照ELISA试剂盒的说明严格操作,最终在Synergy2多功能酶标仪上检测分析。
8. 大鼠脑组织免疫组织化学检测:辐照后在各个观察时间点处死大鼠。断头取出脑组织于10%的甲醛固定液中固定24 h后,常规石蜡包埋,切片,粘片,脱蜡水化,抗原修复,免疫反应,脱水,透明,封片,用光学显微镜观察及图像采集。结果判定:细胞质染色阳性为黄色或黄棕色。每张切片在400倍光镜下随机选择照射区域,3个不重复的视野计数阳性细胞数(不包括阳性的血管内皮细胞),阴性,为0分;阳性细胞数 < 5%,为1分;阳性细胞数6%~15%,为2分;阳性细胞数16%~30%,为3分;阳性细胞数31%~50%,为4分;阳性细胞数>50%,为5分。
9. 统计学处理:数据以 ± s表示。采用SPSS 19.0软件进行分析,两组间比较采用独立样本t检验。P<0.05为差异有统计学意义。
± s表示。采用SPSS 19.0软件进行分析,两组间比较采用独立样本t检验。P<0.05为差异有统计学意义。
1. NF-κB mRNA的表达:采用QRT-PCR方法检测各组大鼠脑组织中NF-κB的基因表达,详细结果列于表2。由结果可知,照射后NF-κB mRNA的表达在第1天就开始增加,受照后第3天达到表达高峰,随后表达量开始减少,至受照后第28天恢复到正常水平。与健康对照组相比,第3、7天升高,差异有统计学意义(t=37.79、35.30,P < 0.05)。
| 表2 QRT-PCR检测辐照后不同时间点脑组织中NF-κB mRNA表达变化( ± s)
|
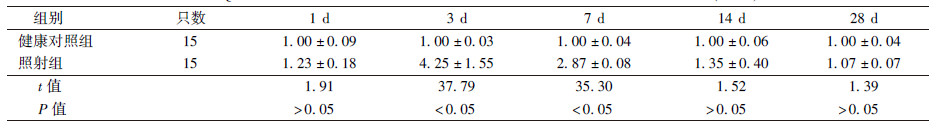
2. NF-κB蛋白质水平的ELISA检测:不同时间点脑组织中NF-κB蛋白变化结果列于表3。与健康对照组比较,大鼠脑组织在照射后第1天,NF-κB蛋白质表达即达到最高峰,随后表达量开始下降,至受照后第28天表达量已基本恢复到正常水平。受照后第1、3、7、14天蛋白表达量与健康对照组相比,差异有统计学意义(t=30.94、14.87、27.17、13.27,P < 0.05)。
| 表3 ELISA检测辐照后不同时间点脑组织中NF-κB蛋白变化(ng/ml, ± s)
|

3. NF-κB蛋白质水平的Western blot检测:结果如图1所示,与ELISA检测结果一致,与健康对照组相比,NF-κB蛋白质水平在受照后第1天达到表达高峰,第3天开始下降,逐渐接近健康对照组水平。

|
图 1 20 Gy电子线作用下不同时间点大鼠脑组织NF-κB蛋白的表达变化 |
4. NF-κB蛋白在海马区域的分布:结果见表4和图2所示,健康对照组NF-κB染色为阴性。在受照后1 d海马区即出现NF-κB染色阳性,背景颜色最深;从受照后第3天开始阳性强度逐渐下降,至受照后第28天已经基本恢复到对照组水平。同时在400倍镜下对NF-κB染色阳性的细胞数进行统计,NF-κB染色阳性细胞个数评分结果列于表4。结果显示,在受照后第1、3、7天 NF-κB染色阳性的细胞数量高于健康对照组,差异有统计学意义(t=-8.49、-4.47、-3.46,P < 0.05)。
| 表4 NF-κB染色阳性细胞个数评分结果( ± s)
|


|
图 2 免疫组织化学法检测急性放射性脑损伤模型中NF-κB蛋白表达情况 ×100 A~F分别是健康对照组、受照后1、3、7、14及28 d海马区域切片 |
放疗在头颈部肿瘤的治疗中有着至关重要的作用,对于生殖细胞瘤、脑干胶质瘤可能是唯一的治疗方法[7]。但放射线是一把"双刃剑",在治疗的同时也不可避免地造成瘤体周围的正常组织的放射性损伤,制约着肿瘤靶区的剂量增加,影响肿瘤的局部控制,导致对放射性脑损伤的防治成为头颈部肿瘤放射治疗中亟待解决的关键问题。本实验采用安全限制内的照射剂量,成功建立放射性脑损伤动物模型,观察研究了NF-κB在放射性脑损伤发生过程中的动态变化规律,为防治放射性脑损伤的发生提供新的思路。
NF-κB通过调节靶基因的转录参与多种细胞信号通路,NF-κB在RBI发生发展过程中所起的作用也成为近年来研究的热点。本研究发现,正常脑组织在接受照射后第1天,NF-κB mRNA的表达就开始增加,到照射后第3天达到高峰。而Western blot及ELSIA方法检测都显示其蛋白质在照射后第1天即达到表达最高峰,高表达一直持续到受照后第28天才恢复到正常水平。同时,本研究还观察到,NF-κB mRNA表达高峰出现的时间滞后于蛋白质表达高峰出现的时间。分析原因可能是,静息状态下,NF-κB二聚体与丝氨酸激酶(Ⅰ-κB)构成三聚体的形式存在细胞质中,当细胞受到电离辐射的刺激后,Ⅰ-κB被磷酸化降解,首先激活NF-κB蛋白质,激活释放NF-κB进入细胞核与相关DNA序列结合启动基因转录[8]。NF-κB的半衰期很短,其下游基因表达的产物反过来又可以维持NF-κB的活性,转录翻译产生的细胞因子,例如细胞间黏附分子-1(ICAM-1)能进一步激活NF-κB从而形成正反馈调节,在炎症持续和扩大中起重要作用[9],这可能是导致NF-κB基因水平表达增高滞后于蛋白质水平的原因。这种正反馈调节可能是促进放射性脑损伤持续发生的重要因素之一。
研究表明,NF-κB活化在脂多糖、缺血、电离辐射等因素诱发的血管内皮细胞损伤中发挥重要的调控作用[10, 11]。本实验结果显示,大鼠脑组织在接受照射后NF-κB基因和蛋白质表达均增加。1995年,Hong等[12]就发现在小鼠脑组织接受照射后,脑组织中的NF-κB被激活。Raju等[13]也发现SD大鼠在接受137Cs单次剂量15 Gy的照射后2 h,大脑皮质中NF-κB活性明显增高。国内学者同样发现在放射性兔脑损伤的组织中,NF-κB的表达量明显高于正常脑组织[14]。由此可见,接受过照射的脑组织中,NF-κB的表达量增高,而活化的NF-κB通过调节多种细胞因子的表达增加,或直接促进放射性脑损伤的发生发展;或促进大量自由基产生,损害神经细胞膜、细胞核,引起DNA断裂导致细胞死亡,活化金属蛋白酶,引发血脑屏障破坏,引起血管源性脑水肿,进一步加重脑损伤程度。
多项研究发现,很多细胞因子及生长因子基因转录活化严格受NF-κB的调控,例如血管内皮生长因子(VEGF)、ICAM-1、肿瘤坏死因子-α(TNF-α)、干扰素-γ(INF-γ)等[15, 16]。而这些因子在放射性脑损伤的发生发展过程中起着重要的作用,VEGF可能是造成放射性脑损伤相关的毛细血管损伤、放射性血管源性水肿的关键因素[17, 18]。Yuan等[19]认为ICAM-1表达上调,可以引起细胞骨架的重排,使得内皮细胞肿胀变形,血管损伤,破坏血脑屏障,加重脑水肿的发生。同时,增加白细胞与内皮细胞的粘附力,堵塞微血管,使得微循环障碍。本课题的前期实验结果显示,对大鼠全脑照射20 Gy电子射线后,形成的放射性脑损伤模型的HE染色病理学观察显示,受照后1 d即出现脑毛细血管间隙增宽,内膜破裂,部分血管出现血栓,受照后3~7 d出现血栓高峰,伴随血管间隙进一步增宽[6],结果与文献报道一致。在放射性脑损伤的发生发展过程中,NF-κB可能通过上调其下游基因的表达,增加相关因子的分泌,来促进放射性脑损伤的发生。
近年来,关于放射治疗导致的认知功能的损害受到越来越多学者的关注,这也成为放射性脑损伤防治的重要方面之一。而这种损害主要表现为海马依赖性的认知功能下降,而非海马依赖的认知功能损害不明显[20]。本研究通过免疫组织化学染色的方法,特异性地观察了海马区NF-κB表达情况,希望进一步探讨海马组织在放射过程中发生损害的作用机制。免疫组织化学结果显示,照射后第1天海马区NF-κB表达增加。表达增加的NF-κB可以激活海马区的小胶质细胞[21]。小胶质细胞可产生炎性因子,如TNF-α和IL-1β等[22, 23]。这些细胞因子可以通过诱导海马神经元祖细胞的凋亡,以及阻滞细胞周期的进展,抑制海马神经元细胞的增殖[24, 25]。由此可见,辐照引起海马区NF-κB的表达增加,进而激活其下游炎性因子,促进神经元的凋亡,同时抑制神经元的增殖,这一机制可能是导致放射性脑损伤的认知功能下降的原因之一。
综合本实验结果及文献报道,可见辐射诱导NF-κB活化,表达增加。NF-κB在RBI的发生过程中起到重要作用,抑制NF-κB的活化,降低其下游基因的表达可能有助于防治放射性脑血管损伤,成为防治RBI发生发展的潜在靶点。
| [1] | Tofilon PJ, Fike JR. The radioresponse of the central nervous system: a dynamic process[J]. Radiat Res, 2000, 153(4): 357-370. |
| [2] | Karin M, Lin A. NF-κB at the cross roads of life and death[J]. Nat Immunol, 2002, 3(3): 221-227. |
| [3] | Bours V, Bonizzi G, Bentires-Alj M, et al. NF-κB activation in response to toxical and therapeutical agents: role in inflammation and cancer treatment[J]. Toxicology, 2000, 153(1-3): 27-38. |
| [4] | Karin M, Cao Y, Greten F, et al. NF-κB in cancer: from innocent bystander to major culprit[J]. Nature Rev Cancer, 2002, 2(4): 301-310. |
| [5] | Liu SF, Malik AB. NF-κB activation as a pathological mechanism of septic shock and inflammation[J]. Am J Physiol Lung Cell Mol Physiol, 2006, 290(4): L622-L645. |
| [6] | 马辰莺,徐晓婷,涂彧,等. VEGF mRNA及蛋白在大鼠放射性脑损伤模型中的动态变化[J]. 中华放射医学与防护杂志, 2014, 34(6): 405-410. |
| [7] | 董晓荣,伍钢. 小胶质细胞和放射性脑损伤[J]. 中华神经医学杂志, 2009,8(6): 646-648. |
| [8] | Latzer J, Papoian GA, Prentiss MC, et al. Induced fit, folding, and recognition of the NF-κB-nuclear localization signals by IκBα and IκBβ[J]. J Mol Biol, 2007, 367(1): 262-274. |
| [9] | Tamanini A, Rolfini R, Nicolis E, et al. MAP kinases and NF-κB collaborate to induce ICAM-1 gene expression in the early phase of adenovirus infection[J]. Virology, 2003, 307(2): 228-242. |
| [10] | Guo F, Zhou Z, Dou Y, et al. GEF-H1/RhoA signalling pathway mediates lipopolysaccharide-induced intercellular adhesion molecular-1 expression in endothelial cells via activation of p38 and NF-κB[J]. Cytokine, 2012, 57(3): 417-428. |
| [11] | Son EW, Rhee DK, Pyo S. γ-irradiation-induced intercellular adhesion molecule-1(ICAM-1) expression is associated with catalase: activation of Ap-1 and JNK[J]. J Toxicol Environ Health A, 2006, 69(24): 2137-2155. |
| [12] | Hong JH, Chiang CS, Campbell IL, et al. Induction of acute phase gene expression by brain irradiation[J]. Int J Radiat Oncol Biol Phys, 1995, 33(3): 619-626. |
| [13] | Raju U, Gumin GJ, Tofilon PJ. NF-κB activity and target gene expression in the rat brain after one and two exposures to ionizing radiation[J]. Radiat Oncol Invest, 1999, 7(3):145-152. |
| [14] | 刘家斌, 李广生, 邓江华,等. 依达拉奉对急性放射性损伤兔脑组织核转录因子-κB表达及丙二醛含量的影响[J]. 临床神经病学杂志, 2013, 26(5): 358-361. |
| [15] | Mason NJ, Artis D, Hunter CA. New lessons from old pathogens: what parasitic infections have taught us about the role of nuclear factor-κB in the regulation of immunity[J]. Immunol Rev, 2004, 201(1): 48-56. |
| [16] | Kesanakurti D, Chetty C, Rajasekhar Maddirela D, et al. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma[J]. Oncogene, 2013, 32(43):5144-5155. |
| [17] | Nonoguchi N, Miyataken S, Fukumoto M, et al. The distribution of vascular endothelial growth factor-producing cells in clinical radiation necrosis of the brain: pathological consideration of their potential roles[J]. J Neurooncol, 2011, 105(2): 423-431. |
| [18] | Chao ST, Ahluwalia MS, Barnett GH, et al. Challenges with the diagnosis and treatment of cerebral radiation necrosis[J]. Int J Radiat Oncol Biol Phys, 2013, 87(3): 449-457. |
| [19] | Yuan H, Gaber MW, McColgan T, et al. Radiation-induced permeability and leukocyte adhesion in the rat blood-brain barrier: modulation with anti-ICAM-1 antibody[J]. Brain Res, 2003, 969(1-2): 59-69. |
| [20] | 高玲,刘青杰. 辐射对海马神经元的影响及其作用机制研究[J]. 中国医学装备, 2013, 10(11): 1-3. |
| [21] | Saponaro C, Cianciulli A, Calvello R, et al. The PI3K/Akt pathway is required for LPS activation of microglial cels[J]. Immunopharm Immunot, 2012, 34(5): 858-865. |
| [22] | Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects[J]. Microsc Res Tech, 2001, 54(1): 47-58. |
| [23] | Ungvari Z, Podlutsky A, Sosnowska D, et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells:role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity[J]. J Gerontol A Biol Sci Med Sci, 2013, 68(12): 1443-1457. |
| [24] | Koo JW, Duman RS. IL-1β is an essential mediator of the antineurogenic and anhedonic effects of stress[J]. Proc Natl Acad Sci USA, 2008, 105(2): 751-756. |
| [25] | Bernardino L, Agasse F, Silva B, et al. Tumor necrosis factor-α modulates survival, proliferation,and neuronal differentiation in neonatal subventricular zone cell cultures[J]. Stem Cells, 2008, 26(9): 2361-2371. |