 2015,Vol. 35
2015,Vol. 35



2. 430023 武汉,华中科技大学同济医学院附属协和医院中心实验室
放射治疗是头颈部原发肿瘤及转移瘤的有效治疗手段,但同时难以避免放射线对肿瘤周围正常组织的损伤,临床上主要表现为急性和迟发性的脑损伤[1]。近年来,抑制小胶质细胞的活化,减轻炎症反应因子的释放,被认为是预防放射性脑损伤的关键措施[2]。参芪扶正注射液主要由黄芪和党参两种成分组成[3]。有研究表明,参芪扶正注射液能够抑制脂多糖(LPS)所诱导的炎性反应,但是该药对放射性脑损伤的具体机制尚不清楚。本实验通过观察参芪扶正对放射性脑损伤后小胶质细胞活化后水平与炎症反应因子的释放抑制作用,初步探讨参芪扶正在急性放射性脑损伤的相关分子机制。
1. 动物与分组:6~8周龄C57BL/6J雄性小鼠144只,体重(25±2.1)g,SPF级,购于武汉大学实验动物中心,许可证号:SYXK(鄂)2010-0057,每笼4只分养,饲养于华中科技大学动物实验中心。实验小鼠按随机数字表法,随机分为空白对照组:不做任何处理;单纯照射组:全脑照射20 Gy;照射后治疗组:全脑20 Gy照射后参芪扶正注射液(20 mg/kg)治疗4周。
2. 照射条件:照射方法参考的文献[4],采用ADAC Pinnacle 3D治疗计划评价脑部等剂量分布,95%的等剂量线包括小鼠的头部。6 MV X射线,吸收剂量率2.0 Gy/min,源皮距MV(SSD)=100 cm,射野40 cm×2 cm,机头180°,组织等效物质(1 cm厚)放置小鼠的头部下方,直线加速器(德国西门子公司)单次后野照射小鼠脑部20 Gy。
3. 组织标本的制备:放疗后3、24、72 h和4周共4个时间点[4],麻醉收集标本。将新鲜脑组织从正中线分开,小鼠的左侧半脑在4%多聚甲醛4℃固定过夜,乙醇二甲苯梯度脱水,石蜡包埋,并以4 μm厚切片,右侧大脑半球-80℃保存。
4. 小鼠空间记忆能力评价:Morris水迷宫方法参照文献[4, 5],由华中科技大学同济医学院公共实验平台提供。各组小鼠接受照射后6周开始实验,记录小鼠入水到登台所需时间作为躲避潜伏期;如果超过60 s未登台,则引导小鼠登台适应30 s,记录潜伏期为60 s。每只小鼠每天进行3次实验,分别从第1、2、4象限池壁中间入水,连续进行5 d,计算各组的平均躲避潜伏期。
5. 测定脑组织TNF-α、IL-1β的变化:按照TRIzol(美国Invitrogen公司)试剂说明书,提取总RNA,加入焦炭酸二乙酸(DEPC)溶解,紫外分光光度计测260、280 nm处的吸光度(A)值,进行样品的浓度测定和质量鉴定。按M-MLV反转录试剂盒(美国Invitrogen公司)说明书进行操作,合成cDNA。目的基因设计引物序列如下:TNF-α的上游引物为5' CCTGTAGCCCACGTCGTAG 3',下游引物为5'GGGAGTAACAAGGTACAACCC 3',扩增产物大小为148 bp;IL-1β的上游引物为5' GAAATGCCACCTTTTGACAGTC 3',下游引物为5' CTGGATGCTCTCAGGACA 3',扩增产物大小为117 bp;GAPDH的上游引物为5'AGGTCGGTGTGAACGGATTTG 3',下游引物为5' GGGGTCGTTGATGGCAACA 3',扩增产物大小95 bp。建立20 μl反应体系。反应条件:热启动95℃ 3 min,95℃ 15 s;58℃ 20 s;72℃ 30 s,在ABI stepone plus RT-PCR仪(美国Applied Biosystems公司)经40个循环后结束反应。相对定量研究放疗后脑组织TNF-α、IL-1β表达。
6. Iba-1免疫荧光:参照文献方法[4],Iba-1兔抗鼠一抗(日本Wako公司)浓度1∶500,切片置于湿盒内4℃过夜;Cy3羊抗兔二抗(美国Invitrogen公司)孵育,浓度均为1∶200,切片置于湿盒内避光37℃孵育60 min,DAPI染核15 min,甘油封片,荧光显微镜(日本Olympus公司)拍照,使用Image-proplus 6.0软件,在高倍视野(×40)计数阳性细胞,计算平均阳性细胞数。
7. 小胶质细胞p65免疫荧光:取对数生长期的细胞,制成悬液混匀,37℃、5%CO2培养24 h,照射前0.5 h弃培养基,PBS漂洗,加入参芪扶正注射液继续培养,24 h后收集细胞,3.7%多聚甲醛固定15 min,PBS洗3次,每次5 min,加入冷的1%Triton X-100,4℃ 15 min;PBS洗3次,每次5 min,5%BSA封闭1 h,加入p65兔抗小鼠(美国CST公司),4℃过夜;Cy3羊抗兔二抗(美国Invitrogen公司)孵育,浓度为1∶200,湿盒内避光37℃孵育60 min,DAPI染核15 min,甘油封片,荧光显微镜(日本Olympus公司)拍照。
8. 统计学处理:数据用x±s表示。采用SPSS 18.0软件进行分析,TNF-α、IL-1β mRNA相对表达量的组间差别用t检验,其余用ANOVA检验。P<0.05为差异有统计学意义。
1. Morris水迷宫实验:与空白对照组比较,在照射后1、2、3 d,差异无统计学意义;4、5 d后照射组躲避潜伏期较对照组延长,而照射后参芪扶正注射液可以明显缩短小鼠躲避潜伏期的时间(t=6.34、6.70,P<0.05,表 1)。
|
|
表 1 X射线照射后不同时间Morris水迷宫实验躲避潜伏期的结果(x±s) |
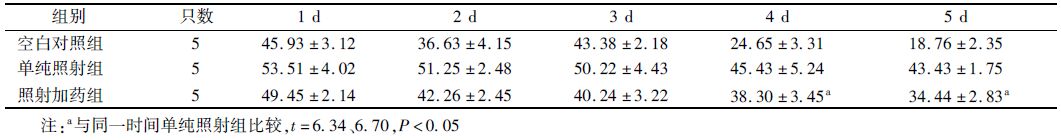
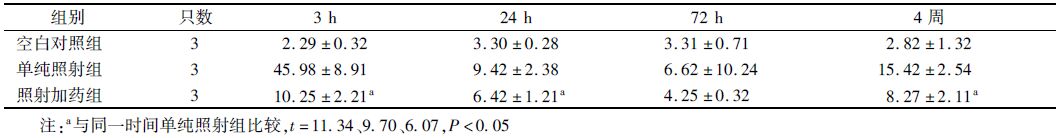
2. 脑组织中TNF-α与IL-1β表达变化:照后3 h TNF-α达到高峰,其后逐渐下降,在4周后又达到第2个高峰,照射后参芪扶正注射液治疗,TNF-α的表达明显下降(t=11.34、9.70、6.07,P<0.05,表 2);照射后IL-1β逐渐升高,在72 h达到高峰,然后逐渐下降。参芪扶正注射液治疗后,IL-1β的表达降低(t=12.27、5.70、7.52,P<0.05,表 3)。
|
|
表 2 X射线照射后不同时间点各组炎症因子TNF-α表达水平(x±s) |
|
|
表 3 X射线照射后不同时间点各组炎症因子IL-1β表达水平(x±s) |

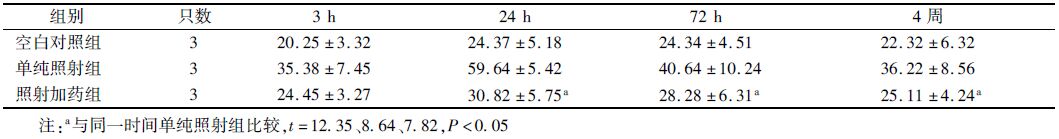
3. 海马区Iba-1免疫荧光:空白对照组中海马区的小胶质细胞较少,单纯照射组中激活状态的小胶质细胞数目明显增多,而照射后参芪扶正注射液治疗后海马区小胶质细胞数目明显降低,差异有统计学意义(t=12.35、8.64、7.82,P<0.05,表 4)。
|
|
表 4 X射线照射后不同时间点各组小胶质细胞数目(x±s) |
4. 小胶质细胞p65免疫荧光:在空白对照中,p65主要表达在小胶质细胞的包膜中,而照射后p65胞膜表达水平降低,而胞核表达水平增加,说明照射后p65在小胶质细胞中存在胞质、胞核的转位,而参芪扶正注射液治疗后可以明显抑制p65的转位(图 1)。
 |
图 1 激光显微镜下观察小胶质细胞p65免疫荧光染色 ×400 注:DAPI.细胞核染料定位细胞;p65.NF-κB的亚基;Merge.融合图 |
放疗是临床上头颈部原发肿瘤以及转移瘤的常见治疗手段,但是在放疗的同时,也不可避免地引起肿瘤周围组织的损伤,其病理学是一个伴随着损伤和修复的动态过程[6]。目前放射性脑损伤的发病机制主要集中在3个方面:血管损伤、胶质细胞损伤和自身免疫反应[7]。一般认为放射性脑损伤的急性期主要表现为以血管为中心的炎性反应,即血管内皮细胞的凋亡,血脑屏障的破坏等病理变化。而炎性细胞主要包括淋巴细胞,少量中性粒细胞及定居脑组织中的小胶质细胞[8]。
本实验成功建立了放射性脑损伤的模型,并在全脑照射后6周,头部照射导致海马齿状回神经发生减弱与空间认知障碍在许多研究中得到证实[4, 9]。Morris水迷宫是目前用于测试实验动物空间位置记忆学习能力的常用方法,并广泛用于神经科学的研究[10],本实验的动物经水迷宫实验检测,小鼠头部全脑放疗后6周,照射组与对照组在躲避潜伏期在第4、5天差异显著,而参芪扶正注射液治疗后小鼠的认知功能明显改善,提示参芪扶正注射液对放射性脑损伤的保护作用可能与海马区神经元细胞损伤减轻有关。
小胶质细胞是CNS定居的巨噬细胞,对各种形式的刺激能产生反应。小胶质细胞表达一系列结构表面分子,包括F4/80,Iba-1,CD11b等。而Iba-1免疫荧光显示,放射性脑损伤后海马区小胶质细胞不同程度的激活。PCR结果显示,在小胶质细胞的激活过程中,伴有炎性因子TNF-α与IL-1β不同程度的增高[11],而参芪扶正注射液治疗组可以明显抑制炎性因子的释放,提示可能与参芪扶正注射液抑制小胶质细胞的激活,从而减轻炎性因子TNF-α与IL-1β对脑组织的损伤作用有关。
PIDD(p53-induced protein with a death domain)作为p53的下游靶基因,在调节细胞凋亡与应急修复方面具有重要作用[11]。PIDD参与活化NF-κB通路,后者可以促进细胞DNA损伤后的修复。静息状态下NF-κB与IκB在细胞质里集合,形成无活性的状态,放射性脑损伤后PIDD-FL和HSP90相互作用,参与PIDD-FL的自身裂解和激活[12],PIDD和Caspase-2以及RAIDD(RIP-associated ICH1/CED3-homologus protein with death domain)形成PIDDosome. 而RIP-1-PIDDosome导致NEMO的磷酸化,激活NF-κB通路,主要表现为小胶质细胞中磷酸化p65胞质胞核转位和TNF-α与IL-1β炎性因子的释放,后者与和Caspase-2结合,促进凋亡能力增加,抑制NF-κB通路的能力[12, 13]。
综上所述,放疗小胶质细胞活化,激活NF-κB p65胞质胞核转位,导致炎性因子TNF-α与IL-1β的释放,最后导致放射性脑损伤。参芪扶正注射液对放射性脑损伤的保护作用分子机制,可能与抑制NF-κB p65胞质胞核转位,减轻炎症因子的产生,减轻放射诱导的脑损伤有关。
| [1] | Khuntia D, Brown P, Li J, et al. Whole-brain radiotherapy in the management of brain metastasis[J]. J Clin Oncol, 2006, 24(8): 1295-1304. |
| [2] | Zhu C, Huang Z, Gao J, et al. Irradiation to the immature brain attenuates neurogenesis and exacerbates subsequent hypoxic-ischemic brain injury in the adult[J]. J Neurochem, 2009, 111(6): 1447-1456. |
| [3] | Dong XR, Wang JN, Liu L, et al. Modulation of radiation-induced tumour necrosis factor-alpha and transforming growth factor beta1 expression in the lung tissue by Shengqi Fuzheng injection[J]. Mol Med Rep, 2010, 3(4): 621-627. |
| [4] | Dong X, Luo M, Huang G, et al. Relationship between irradiation-induced neuro-inflammatory environments and impaired cognitive function in the developing brain of mice[J]. Int J Radiat Biol, 2015, 91(3):224-239. |
| [5] | Inostroza M, Cid E, Brotons-Mas J, et al. Hippocampal-dependent spatial memory in the water maze is preserved in an experimental model of temporal lobe epilepsy in rats[J]. PLoS One, 2011, 6(7): e22372. |
| [6] | Wang S, Qiu D, So KF, et al. Radiation induced brain injury: assessment of white matter tracts in a pre-clinical animal model using diffusion tensor MR imaging[J]. J Neurooncol, 2013, 112(1): 9-15. |
| [7] | 黄国栋,罗鸣,屈晓飞,等. 电离辐射抑制海马区神经发生与放射性脑损伤认知损害的关系[J]. 中华放射医学与防护杂志, 2013, 33(2): 113-122. |
| [8] | Xue J, Dong JH, Huang GD, et al. NF-κB signaling modulates radiation-induced microglial activation[J]. Oncol Rep, 2014, 31(6): 2555-2560. |
| [9] | Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair[J]. Nature, 1998, 391(6665): 401-404. |
| [10] | Haber JE. DNA recombination: the replication connection[J]. Trends Biochem Sci, 1999, 24(7):271-275. |
| [11] | Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects[J]. Microsc Res Tech, 2001, 54(1): 47-58. |
| [12] | Tinel A,Janssens S, Lippens S,et al. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and prosurvival NF-kappaB pathway[J]. EMBO J, 2007, 26(1): 197-208. |
| [13] | Ando K, Kernan JL, Liu PH, et al. PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling[J]. Mol Cell, 2012, 47(5): 681-693. |