 2015, Vol. 35
2015, Vol. 35



辐射能诱发细胞基因组不稳定性,导致细胞正常复制过程失去平衡,受照的亲代细胞将基因组不稳定性传递给子代的同时,还增加了癌变发生的风险性[1]。基因组不稳定性是癌症细胞的标志,被认为与致癌过程相关[2]。因此,研究基因组不稳定性可以深入了解辐射致癌的发生发展,并为癌症的诊断及治疗提供新的靶点。本课题组前期研究表明,电离辐射可诱发人正常肝细胞基因组不稳定的发生[3],基因芯片检测发现CDT1基因在不同剂量照射后的人正常肝细胞HL-7702子代中差异表达显著[4],推测CDT1基因活性的调节对于维持基因稳定性具有非常重要的作用。因此,本实验利用慢病毒介导的过表达技术上调CDT1基因的表达,放大可能的基因组不稳定性作用效果,观察CDT1基因上调后对基因组不稳定肝细胞模型HL-7702细胞周期及凋亡的影响。
1. 细胞系、主要试剂及仪器:辐射诱发的基因组不稳定肝细胞HL-7702模型由本实验室前期构建、保存[3]。RPMI 1640培养基(美国GIBCO公司),胎牛血清(天津市灏洋生物制品公司),过表达慢病毒(上海吉凯基因化学公司),细胞DNA含量检测试剂盒、Annexin V-APC/7-AAD细胞凋亡检测试剂盒(南京凯基生物公司),RNAprep pure培养细胞/细菌总RNA提取试剂盒(北京天根生化公司),PrimeScriptTM RT试剂盒及实时荧光定量PCR试剂SYBR Premix Ex TaqTM Ⅱ(大连宝生物工程公司),流式细胞仪(美国BD公司)。
2. 慢病毒的构建及感染:针对CDT1基因设计并合成引物,上游引物:5' GGACTCGTGCTGCCCTACAA 3',下游引物:5' CCTCCTGGTGCCATCCTTG 3'。以人肝细胞基因组CDT1基因cDNA为模板,构建并包装慢病毒GV218-CDT1。同时,包装含有随机序列的慢病毒作为阴性病毒GV218-NC。GV218-CDT1及GV218-NC病毒滴度分别为4.3×108和2×109 TU/ml。慢病毒包装由上海吉凯基因化学技术有限公司协助完成。
感染时接种细胞数约为5×104的基因组不稳定HL-7702细胞于24孔板,待培养细胞融合度达到30%时,过表达慢病毒GV218-CDT1与阴性对照慢病毒GV218-NC分别感染基因组不稳定HL-7702细胞,每孔感染复数(感染时病毒与细胞数量的比值,MOI)=50,polybrene 5 μg/ml,振荡混匀后37℃、5% CO2继续培养。分为慢病毒感染组GV218-CDT1和阴性慢病毒感染组GV218-NC。感染前使用无血清培养基,于12 h更换20%胎牛血清培养基,感染后3 d,在荧光显微镜下观察绿色荧光蛋白(GFP)的表达情况。待GFP荧光数量表达稳定后收集细胞,用于后续检测。
3. 流式细胞术检测细胞周期:慢病毒感染细胞后,待细胞中GFP荧光表达稳定后收集细胞,每个样品收集2×106个细胞,用PBS洗涤细胞并去除上清,加入 1 ml 70%的冷乙醇固定过夜,后用PBS洗涤,加入100 μl RNase,200 μl PI染色混匀,避光30 min后上机,1 h内检测。每个实验组设3个平行样品。
4. 流式细胞术检测细胞凋亡:慢病毒感染细胞后,待细胞中GFP荧光表达稳定后收集细胞,每个样品收集2×105个细胞。用1 ml预冷的PBS洗涤细胞2次,4℃离心,离心加速度206×g,1 500 r/min,离心5 min,加入4 μl Annexin V-APC,混匀后加入4 μl 7-AAD,混匀,室温避光反应15 min,上机前加入250 μl的Binding Buffer,1 h内利用流式细胞仪进行细胞凋亡率检测。每个实验组设3个平行样品。
5. CDT1基因过表达对p53、ATM、ATR、Bcl-2和Caspase-3基因mRNA表达的影响:慢病毒感染细胞后,荧光显微镜下观察,待GFP表达稳定后,收集感染细胞,提取总RNA,反转录cDNA,利用实时荧光定量PCR (RT-PCR) 分析CDT1基因过表达对p53、ATM、ATR、Bcl-2和Caspase-3基因mRNA表达变化的影响。RT-PCR的方法检测p53、ATM、ATR、Bcl-2、Caspase-3基因的表达情况,反应条件为95℃ 10 s;95℃ 5 s,60℃ 20 s(40个循环),以ΔΔCt法进行相对定量分析各个基因mRNA水平。各基因引物序列见表 1。
| 表 1 引物序列 |

6. 统计学处理:实验数据以x±s表示。采用SPSS Statistics V21.0软件进行分析。两组间差异采用独立样本t检验。P<0.05为差异有统计学意义。
1. 慢病毒介导的过表达效果检测:慢病毒感染组不稳定肝细胞3 d后在荧光显微镜下可观察到绿色荧光蛋白GFP的表达,见图 1。流式细胞术检测慢病毒感染效率达到65%。RT-PCR检测表明,阴性慢病毒转染组CDT1基因表达量为0.76±0.14,慢病毒转染组CDT1基因表达量为10.31±1.73。与阴性慢病毒感染组相比,慢病毒感染组CDT1基因mRNA表达水平明显升高(t=15.56,P<0.05),CDT1基因过表达效果较好。
 | 图 1 慢病毒感染基因组不稳定肝细胞荧光显微镜观察×100 A.相差显微镜观察;B.荧光显微镜同一视野观察 |
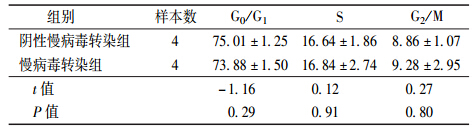
2. CDT1过表达对基因组不稳定肝细胞细胞周期的影响:PI单染流式细胞术检测基因组不稳定肝细胞细胞周期结果显示,两转染组细胞各细胞周期比例差异无统计学意义(表 2)。
| 表 2 CDT1过表达对两转染组细胞周期的影响(%,x±s) |
3. CDT1过表达对基因组不稳定肝细胞细胞凋亡的影响:Annexin V-APC流式细胞术检测基因组不稳定细胞凋亡情况,结果显示,阴性慢病毒转染组细胞凋亡率为(0.71±0.23)%,慢病毒转染组凋亡率为(1.48±0.30)%,差异有统计学意义(t=4.19,P<0.05)。表明CDT1过表达能够促进基因组不稳定肝细胞的凋亡。
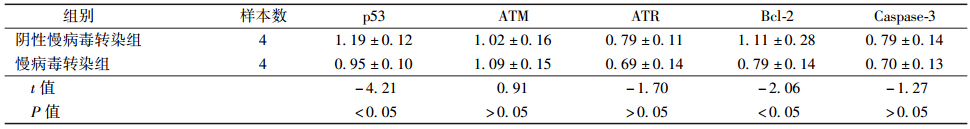
4. CDT1过表达对基因组不稳定肝细胞p53、ATM、ATR、Bcl-2、Caspase-3基因表达的影响:阴性慢病毒转染组靶基因表达量为1,慢病毒转染组样品靶基因定量计算,实时荧光定量PCR检测结果表明,CDT1过表达可使基因组不稳定肝细胞p53、Bcl-2基因mRNA转录水平下调(t=-4.21、-2.06,P <0.05) 。ATM基因mRNA转录水平上调,ATR、Caspase-3基因mRNA转录水平下调,但差异均无统计学意义(表 3)。
| 表 3 CDT1过表达对两转染组p53、ATM、ATR、Bcl-2、Caspase-3基因表达的影响(x±s) |
Cdt1是CDT1基因(Cdc10 dependent transcript 1,Cdc10依赖性转录因子1)编码的蛋白,是在基因复制调控中起重要作用的蛋白因子,参与构成复制许可因子,是DNA复制起始必不可少的成分。复制许可因子保证每个细胞周期基因组只复制一次,从而维持基因组稳定[5, 6]。Chipuk等[7]在H1299肺癌细胞中发现,CDT1的过度表达足以造成在同一个细胞周期中细胞的重复复制。因此,CDT1基因的过表达可能造成基因组不稳定性的发生。本实验利用辐射诱发的基因组不稳定性肝细胞过表达CDT1基因,放大可能的基因组不稳定性作用效果,从而研究CDT1基因对辐射诱发基因组不稳定的作用。
基因组不稳定性首次由Kadhim等[8]提出,表现为亲代辐射暴露后子代细胞中存在较高的细胞异常,且这种改变具有遗传性。为防止损伤随着细胞的分裂不断向下传递,最终出现突变表型,细胞往往会出现细胞周期阻滞,因此,细胞周期阻滞是基因组稳定性的重要维护途径[9]。ATM及其相关激酶ATR被认为是细胞周期检查点信号通路的主要控制器,细胞受损后ATM参与的一系列磷酸化促使细胞周期阻滞[10]。然而,本实验结果显示,两转染组相比,细胞周期均无明显阻滞,而且,ATM、ATR基因在mRNA水平亦差异无统计学意义。有研究证实,哺乳动物细胞中,电离辐射诱导的CDT1水解存在不依赖于ATM-Chk2和ATR-Chk1通路[11]。推断是由于出现检查点的适应性,在DNA损伤或损伤未修复时,细胞以一种替代途径进入细胞周期。有文献报道,检查点的适应性是促进基因组不稳定性和致癌的潜在因素[12]。
当无法正确修复的DNA累积到一定程度,凋亡相关基因被激活诱导细胞凋亡,从而维持基因组稳定性。实验结果显示,辐射诱发的基因组不稳定性肝细胞过表达CDT1基因后,凋亡率显著高于阴性转染组,且p53出现显著下调。文献报道存在非p53依赖的凋亡途径,Lackinger等[13]研究发现,在p53缺失时,DNA损伤可诱导细胞凋亡。如在 DNA损伤剂诱导下,p53敲除的小鼠成纤维细胞能够发生凋 亡,且凋亡程度与野生型细胞相似。为进一步验证CDT1基因过表达与凋亡的关系,揭示CDT1基因过表达触发非p53依赖的凋亡途径,本研究检测了CDT1基因过表达对Bcl-2、Caspase-3基因的影响。Caspase-3是p53依赖的凋亡途径中重要的 执行蛋白,以p53依赖的途径发挥作用[14],Bcl-2是p53下游的凋亡相关基因中的抑凋亡基因,与p53基因相互抑制调节凋亡[15]。实验结果显示,Bcl-2、Caspase-3基因表达下调。因此,推测CDT1基因过表达通过诱导一种不依赖p53的途径的凋亡,参与基因组不稳定肝细胞凋亡的调控。
综上所述,CDT1过表达可能通过下调p53、ATR、Bcl-2、Caspase-3基因及上调ATM基因,参与基因组不稳定肝细胞周期及凋亡的调控,促进基因组不稳定肝细胞的凋亡,提示CDT1基因过表达通过参与非p53依赖的凋亡途径以及检查点适应性对基因组不稳定性进行调控。
| [1] | Vorobtsova IE. Transgenerational transmission of radiation-induced genomic instability and predisposition to garcinogenesis[J]. Vopr Onkol, 2008,54(4):490-493. |
| [2] | Kaup S,Grandjean V, Mukherjee R, et al. Radiation-induced genomic instability is associated with DNA methylation changes in cultured human keratinocytes[J]. Mutat Res,2006, 597(1-2):87-97. |
| [3] | Zuo YH, Dang XH, Zhang HF, et al. Genomic instability induced by ionizing radiation in human hepatocytes[J]. J Toxicol Environ Health A,2012,75(12):700-706. |
| [4] | 左雅慧,党旭红,王放,等. 60Co γ射线照射后人正常肝细胞子代的基因差异表达[J]. 中华放射医学与防护杂志,2011,31(4):425-429. |
| [5] | Wohlschlegel JA, Dwyer BT, Dhar SK, et al. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1[J]. Science, 2000, 290(5500):2309-2312. |
| [6] | Vaziri C, Saxena S, Jeon Y, et al. A p53-dependent checkpoint pathway prevents rereplication[J]. Mol Cell, 2003, 11(4): 997-1008. |
| [7] | Chipuk JE, Maurer U, Green DR, et al. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription[J]. Cancer Cell, 2003, 4(5): 371-381. |
| [8] | Kadhim MA, Macdonald DA, Goodhead DT, et al. Transmission of chromosomal instability after plutonium alpha-particle irradiation[J]. Nature,1992,355(6362):738-740. |
| [9] | Ning S, Knox SJ. G2/M-phase arrest and death by apoptosis of HL60 cells irradiated with exponentially decreasing low-dose-rate gamma radiation[J]. Radiat Res, 1999, 151(6): 659-669. |
| [10] | 郭海卓.辐射诱导人淋巴细胞DNA损伤及细胞周期调控基因表达的量效关系[D].长春:吉林大学,2010. |
| [11] | Truong LN, Wu X. Prevention of DNA re-replication in eukaryotic cells[J]. J Mol Cell Biol, 2011,3(1):13-22. |
| [12] | Swift LH, Golsteyn RM. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells[J]. Int J Mol Sci, 2014,15(3):3403-3431. |
| [13] | Lackinger D, Eichhorn U, Kaina B, et al. Effect of ultraviolet light, methyl methanesulfonate and ionizing radiation on the genotoxic response and apoptosis of mouse fibroblasts lacking c-Fos, p53 or both [J]. Mutagenesis,2001,16(3): 233-241. |
| [14] | 钱呈睿,葛海良,王颖.p53转录非依赖活性介导细胞凋亡[J].生命科学,2007,19(3):326-329. |
| [15] | 王文文,邓毛程. Bcl-2、Bax及Bid与细胞周期阻滞及凋亡调控的关系[J].广东轻工职业技术学院学报,2009,8(3):21-24. |