 2015, Vol. 35
2015, Vol. 35



长期以来,心脏被认为是一个对放射线抵抗的器官。然而实验表明,经历了放疗的早期乳腺癌患者的生存获益几乎被心血管不良反应抵消[1]。虽然放疗技术的进展减少了心脏的受照射范围及剂量,但胸部放疗时,小部分心脏始终能接受到高剂量,或是整个心脏能接受到较低剂量的射线[2],可能导致放射性心脏损伤(RIHD)。RIHD发生的机制是血管及心肌的纤维化,所以抑制射线所致的纤维化过程可能减轻RIHD。过氧化物酶体增生物激活受体-γ (PPAR-γ) 是配体激活转录因子细胞核受体超家族的一员,通过两种方式发挥作用,反式激活 (主要导致代谢效应) 及反式阻抑(主要发挥抗炎效果) [3]。吡格列酮(Pio)是PPAR-γ的选择性激动剂,通过抑制黏附分子及炎症因子的表达发挥抗炎作用,也能通过抑制转化生长因子β1 (TGF-β1)信号通路来发挥抗纤维化作用[4, 5]。许多研究表明,PPAR-γ激动剂能抑制许多器官的纤维化,如肺及心血管系统[6, 7]。但目前为止,少见报道吡格列酮的放射性心脏保护作用。因此,本研究设想吡格列酮能减轻RIHD,探讨其是否能减轻大鼠放射性心肌纤维化。
1.实验动物与试剂:SPF级的Sprague-Dawley(SD)大鼠 (中国医科大学动物实验中心) 46只,雌雄各半,3月龄,体重178~245 g(平均219.4 g),合格证号:SCXK(京)2009-0015。大鼠在SPF级实验室饲养,温度20~25℃,湿度50%~55%。实验已获得中国医科大学动物伦理委员会的批准。马松染色试剂盒购自上海博古生物有限公司。PPAR-γ多克隆抗体购自美国Santa Cruz公司;Tris、APS、SDS、TEMED、Tween-20、丙烯酰胺、甲叉双丙烯酰胺、丽春红购自美国Sigma公司;SuperECL Plus超敏发光液、显影液及定影液购自北京尚柏生物技术有限公司。RNA裂解液TRIzol 20 ml×2、cDNA反转录试剂盒、SYBR Premix Ex Taq购自日本TaKaRa公司。
2.动物分组与照射:应用随机数字表法分为6组,每组雌雄数目相等。健康对照组、吡格列酮10 mg·kg-1·d-1组、吡格列酮20 mg·kg-1·d-1组各6只,18 Gy+安慰剂(蒸馏水)组8只,18 Gy照射+吡格列酮10 mg·kg-1·d-1组10只,18 Gy照射+吡格列酮20 mg·kg-1·d-1组10只。健康对照组不给予任何干预,照射组及照射+吡格列酮组,大鼠按照0.3 ml/100 g给予水合氯醛腹腔注射麻醉,麻醉后心脏定位,置于直线加速器下照射(德国Siemens公司Oncor-H型)。使用6 MV高能X射线,前后对穿照射,源皮距100 mm,照射野2 cm×2 cm,吸收剂量率300 cGy/min,射线照射心脏,其余部位由铅板遮挡。心脏接受18 Gy剂量的照射相当于通常用于患者分次照射2 Gy×30次的效果[8]。
3.药物干预与取材:照射完后6 h之内,除健康对照组外,对所有大鼠行第1次灌胃处理,照射组灌入2 ml蒸馏水,吡格列酮及照射+吡格列酮组灌入相应量的吡格列酮。此后每天灌胃1次,每5天测量1次体重,根据体重调整灌药量,行灌胃处理30 d,分笼饲养至3个月。照射后3个月标本取材,用水合氯醛过量麻醉法处死大鼠,取其心脏组织,洗净,用剪刀将其横断,一半迅速放于4%中性甲醛固定液中固定,48 h后转入70%乙醇中保存;另一半-80℃保存。
4.马松染色观察心肌纤维化程度:将保存于70%乙醇中的心肌组织取出,取5 mm×5 mm×5 mm的心脏横断面组织,制成组织蜡块,切成厚度约4 μm的组织切片,烘干,脱蜡。马松复合液染色5 min,0.2%醋酸水溶液洗10 s,5%磷钨酸10 min,0.2%醋酸水溶液浸洗2次,2%苯胺蓝液染色5 min,0.2%醋酸水溶液洗2次,脱水,封片。在光学显微镜下观察心肌细胞等损伤情况和胶原的改变,并拍照。
5. Western blot分析各组蛋白表达:切取0.1 g组织样本,制成组织匀浆,12 000×g离心10 min,取上清。BCA蛋白定量试剂盒(北京尚柏生物技术有限公司)测定蛋白浓度。心肌蛋白由8%的SDS聚丙烯酰胺凝胶电泳分离,然后转移到硝酸纤维素膜上。膜分别由特异抗PPAR-γ和TGF-β1氨基酸抗体孵育(用3%的封闭液1∶500 稀释,美国Santa Cruz公司),CDK5做同样处理(CDK5与缓冲液1∶500稀释,美国Proteintech Group公司)。GAPDH被用作内参 (1∶15 000,上海康成公司,1×TBS-T缓冲液)。洗完膜后,在5%的缓冲液中由二抗孵育(1∶2 000)。信号使用电化学发光(ECL)显色液曝光。用LabWorks软件对图像进行灰度分析。数据用蛋白灰度值/GAPDH蛋白灰度值表示。
6.Real-time PCR观察PPAR-γ mRNA表达情况:通过美国国立图书馆Medline数据库检索PPAR-γ基因序列,应用Primer5.0软件设计,由北京尚柏生物技术有限公司合成引物。上游:5′ ATCGCACTTTGGTATTCTT 3′,下游:5′ CGGTTGATTTCTCCAGCAT 3′。β-肌动蛋白(美国Santa Cruz公司)用作内参。总RNA分离方法参照文献[9],cDNA反转录的过程参照文献[10]。cDNA放入Real-time PCR扩增仪中进行扩增,反应条件为:预变性95℃ 10 s;PCR反应95℃ 5 s,60℃ 20 s,共45个循环;融解曲线分析95℃ 10 s,65℃ 15 s,95℃10 s。记录扩增的内参和目的基因的Ct值及融解曲线,通过2-△△Ct计算目的基因扩增情况。
7.统计学处理:数据以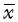 ±s表示,用SPSS 17.0软件分析。组间比较用ANOVA分析,正态资料组间用LSD-t检验。P<0.05为差异有统计学意义。
±s表示,用SPSS 17.0软件分析。组间比较用ANOVA分析,正态资料组间用LSD-t检验。P<0.05为差异有统计学意义。
1.照射后大鼠心脏的病理改变:最终有44只大鼠完成实验,照射+Pio 10 mg·kg-1·d-1组有1只大鼠在照射后第2天死亡,照射+安慰剂组有1只大鼠死亡。照射+安慰剂组大鼠出现食欲不振,毛色暗淡,体重下降明显等症状,健康对照组大鼠无死亡。在200倍镜下观察,心肌马松染色结果示健康对照组,单纯Pio 10 mg·kg-1·d-1组及Pio 20 mg·kg-1·d-1组的大鼠心肌细胞形态正常,无纤维化表现(图1A,B,C),照射+安慰剂组大鼠心肌横切面可见正常的心肌细胞被大量的胶原细胞所替代,心肌内胶原组织明显增多,粗大胶原纤维相互连接成网状,排列紊乱,分布不匀(图1F),而照射+Pio 10 mg·kg-1·d-1组及照射+Pio 20 mg·kg-1·d-1组的纤维化明显比照射+安慰剂组轻(图1D,E)。

|
图1 各处理组大鼠心肌镜下观察 马松染色 ×200 A.健康对照组;B. Pio 10 mg·kg-1·d-1组;C. Pio 20 mg·kg-1·d-1组;D.照射+Pio 10 mg·kg-1·d-1组;E.照射+Pio 20 mg·kg-1·d-1组;F.照射+安慰剂组 |
2.心肌PPAR-γ蛋白表达:Western blot显示,在相对分子质量50 000~55 000附近有明显蛋白表达条带,PPAR-γ蛋白表达在照射组高于未照射组。健康对照组、Pio 10 mg·kg-1·d-1组、Pio 20 mg·kg-1·d-1组、照射+Pio 10 mg·kg-1·d-1组、照射+Pio 20 mg·kg-1·d-1组、照射+安慰剂组目的内参比分别为0.301±0.044、0.352±0.032、0.392±0.038、0.691±0.055、0.732±0.046、0.774±0.061。受照射的3组大鼠PPAR-γ蛋白表达明显高于未照射的3组,差异有统计学意义(F=12.435,P<0.05),见图2。

|
注:1.健康对照组; 2. Pio 10 mg·kg-1·d-1组; 3. Pio 20 mg·kg-1·d-1组; 4.照射+Pio 10 mg·kg-1·d-1组; 5.照射+Pio 20 mg·kg-1·d-1组; 6.照射+安慰剂组 图2 各组大鼠心肌细胞PPAR-γ蛋白的表达水平 |
3. 心肌 PPAR-γ的mRNA表达:6组的2-△△Ct值的比较显示,PPAR-γ mRNA表达趋势与蛋白表达趋势不完全一致,只有照射+Pio 20 mg·kg-1·d-1组与单纯Pio 20 mg·kg-1·d-1组之间差异有统计学意义(F=2.333,P<0.05),其余各组间比较未见差异有统计学意义(P>0.05)。
4. 心肌TGF-β1和Cdk5蛋白表达:Western blot分析显示,TGF-β1与Cdk5分别在相对分子质量12 500和33 000出现明显的条带(图3)。健康对照组、Pio 10 mg·kg-1·d-1组、Pio 20 mg·kg-1·d-1组、照射+Pio 10 mg·kg-1·d-1组、照射+Pio 20 mg·kg-1·d-1组、照射+安慰剂组TGF-β1目的内参比分别为0.173±0.020、0.167±0.023、0.183±0.040、0.201±0.028、0.209±0.026、0.321±0.040。TGF-β1在照射+安慰剂组明显高于其余5组(F=17.578,P<0.05);Cdk5目的内参比分别为0.128±0.027、0.120±0.021、0.131±0.026、0.148±0.022、0.159±0.018、0.185±0.034。Cdk5的表达则在受照射的大鼠中均有升高,而只有照射+安慰剂组的升高差异有统计学意义(F=3.651,P<0.05)(图4)。

|
注:1.健康对照组; 2. Pio 10 mg·kg-1·d-1组; 3. Pio 20 mg·kg-1·d-1组; 4.照射+Pio 10 mg·kg-1·d-1组; 5.照射+Pio 20 mg·kg-1·d-1组; 6.照射+安慰剂组 图3 各组大鼠心肌细胞TGF-β1蛋白的表达水平 |

|
注:1.健康对照组; 2. Pio 10 mg·kg-1·d-1组; 3. Pio 20 mg·kg-1·d-1组; 4.照射+Pio 10 mg·kg-1·d-1组; 5.照射+Pio 20 mg·kg-1·d-1组; 6.照射+安慰剂组 图4 各组大鼠心肌细胞Cdk5蛋白的表达水平 |
Boerma等[8]认为,尽管大鼠与人类相比不易于发生动脉粥样硬化,但在大鼠心脏接受局部照射后,其心肌重构与在人类胸部放疗几年后观察到的相似,因此,大鼠模型能很好地用来研究放射性心脏重构。吡格列酮的剂量选择参考了其在缺血性心脏损伤及心室重构中的作用研究[11, 12]。Smeets等[13]发现,PPAR-γ激动剂的应用时机十分重要,当其开始应用是在永久性冠状动脉堵塞7 d以后,PPAR-γ激动剂并不能影响大鼠的梗阻范围、左室重塑及死亡率,但在心肌梗死开始24 h后给予治疗时,吡格列酮能显著改善血流动力学及减轻左室重塑。到目前为止,没有相关研究表明是否吡格列酮越早应用(24 h内),就能越好地保护心脏。因此,本研究在照射后6 h内第1次给予吡格列酮,而放疗后何时给予PPAR-γ激动剂能更好地保护心脏有待进一步研究。
RIHD的一个显著机制是血管壁及心肌的纤维化,在纤维化过程中,TGF-β的上调及血管紧张素醛固酮(RAAS)系统的激活扮演着重要作用[14, 15]。有研究表明,PPAR-γ配体能抑制TGF-β诱发的皮肤成纤维细胞平滑肌肌动蛋白及胶原Ⅰ的表达[16],而另一个研究表明,PPAR-γ激活能减少Ⅰ型胶原的合成及分泌,这可能与TGF-β1依赖机制有关[17]。噻唑烷二酮类也被报道能减轻经历缺氧-再氧合或用血管紧张素Ⅱ(AngⅡ)处理的人心脏成纤维细胞的增殖,并且能减少Ⅰ型胶原及MMP1的表达,可能是通过抑制氧化应激及NF-κB的激活[18]。有研究发现,PPAR-γ配体能减轻血管紧张素Ⅱ1型受体(AT1R)的表达[19]。最近的研究表明,吡格列酮能通过下调AT1R的表达减轻AngⅡ 诱发心肌成纤维细胞的胶原合成[20]。与这些发现一样,Hao等[21]发现另一种噻唑烷二酮类罗格列酮能抑制AngⅡ诱导的细胞增殖及Ⅰ型、Ⅲ型胶原及纤连蛋白和纤溶酶原激活物1的表达。
前期研究发现,在大鼠心脏接受局部照射后,心肌及血管内皮细胞的PPAR-γ表达增加,然而这种增加没有反式抑制MMP-1、TIMP-1及TGF-β1的表达,因此,心脏的纤维化没有减轻[22]。认为可能的机制是PPAR-γ本身有保护作用,但是在没有予以激动剂时,内源性PPAR-γ蛋白的增加不足以抑制纤维化因子的活性,或者是射线能引起某些抑制PPAR-γ活性的因子表达增加。Choi等[23]研究表明,细胞周期蛋白依赖激酶5(Cdk5)能在PPAR-γ第273位丝氨酸上磷酸化PPAR-γ,同时Cdk5对PPAR-γ的磷酸化能被PPAR配体、罗格列酮及MRL24阻断。就像其他CDKs一样,单体Cdk5不表现出酶活性,并且需要与调节配体结合后才能发挥出活性。目前为止,Cdk5有两个配体被发现,分别是p35和p39,P35/P25(P35的一种有208个残基的羧基端蛋白水解产物) 是许多细胞因子及促炎信号的靶点[24]。Choi等[23]也发现,用TNF-α,IL-6或者游离脂肪酸类处理的3T3-L1 脂肪细胞能引起PPAR-γ第273位丝氨酸的磷酸化。Cdk5在神经系统中表达最高且有最高的激酶活性,能在所有的组织中表达[24]。因此,认为上调的PPAR-γ没有减轻心肌纤维化的原因可能是射线上调了Cdk5 的水平。所以,本实验用PPAR-γ激动剂吡格列酮处理心脏受照射的大鼠,发现吡格列酮能减轻心肌纤维化,且在受照的大鼠心脏中Cdk5表达上调。此外,TGF-β1也在照射+安慰机组明显升高,然而当予以吡格列酮时,对比安慰机组TGF-β1的表达下调。
本研究表明,当大鼠的心脏暴露于18 Gy射线3个月后,心肌的纤维化非常明显,Cdk5、PPAR-γ及TGF-β1的表达也明显上调。然而,当予以受照大鼠吡格列酮1个月后,无论10或是20 mg·kg-1·d-1,心肌的纤维化都减轻,TGF-β1表达的上调也被抑制。所以得出结论,吡格列酮可能通过它的抗纤维化作用,减轻大鼠的放射性心脏损伤,通过抑制Cdk5介导的PPAR-γ的磷酸化起作用。然而,PPAR-γ蛋白表达与mRNA表达的不一致,经过查阅大量文献发现,在一些研究之间也存在不一致。有研究表明,3T3-L1脂肪细胞的PPAR-γ激活能引起PPAR-γ mRNA下调[25];而在另一些研究中,大鼠心肌成纤维细胞及血管用外源性AngⅡ处理后,PPAR-γ的表达无变化,而PPAR-γ的DNA结合能力变小[26, 27, 28],所以,需要更大的样本量来解释这种不一致。配体激活的PPAR-γ与视黄醇X受体形成一个异源二聚体,结合到一个特殊的DNA序列,PPAR反应原件(PPRE),从而激发基因转录[29],称这个效应为“genomic”效应,这个过程需要较长时间。然而,Takeda等[30]发现核受体的配体有迅速的效应,称为“nongenomic”效应,即吡格列酮有一种不依赖PPAR-γ而发挥活性的行为,而依赖磷酸化的细胞外信号调节激酶(ERK)及线粒体5′-AMP激活性蛋白激酶(AMPK)活性。对于PPAR-γ表现出反式激活及反式阻抑的两种活性,是否通过“nongenomic”起效的,尚不明确。本研究中,假设Pio是通过依赖PPAR-γ途径(“genomic”效应)发挥心脏保护作用的。本实验不足之处是没有同时用PPAR-γ敲除的大鼠进行实验。在以后的研究中,将会用PPAR-γ拮抗剂及Cdk5抑制剂来验证假说,希望对RIHD的预防及治疗有帮助。
志谢 感谢中国医科大学附属盛京医院肿瘤科及实验中心的支持,感谢盛京医院放疗科肖福大教授的技术支持
| [1] | Early Breast Cancer Trialists' Collaborative Group. Effects of radiotherapy and surgery in early breast cancer--An overview of the randomized trials[J]. N Engl J Med, 1995,333(22):1444-1455. |
| [2] | Boerma M. Experimental radiation-induced heart disease: Past, present, and future[J]. Radia Res, 2012,178(1):1-6. |
| [3] | Abdelrahman M, Sivaraja A, Thiemerman CH. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock[J]. Cardiovasc Res,2005,65(4):772-781. |
| [4] | Zhao C, Chen W, Yang L, et al. PPAR-gamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells[J]. Biochem Biophys Res Commun, 2006,350(2):385-391. |
| [5] | Chen MM, Lam A, Abraham JA, et al. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis[J]. J Mol Cell Cardiol, 2000,32(10):1805-1819. |
| [6] | Hart CM, Roman J, Reddy R, et al. PPAR gamma: a novel molecular target in lung disease[J]. J Investig Med,2008,56(2):515-517. |
| [7] | Das SK, Chakrabarti R. Role of PPAR in cardiovascular diseases[J]. Recent Pat Cardiovasc Drug Discov,2006,1(2):193-209. |
| [8] | Boerma M, Wang JR, Wondergem J, et al. Influence of mast cells on structural and functional manifestations of radiation-induced heart disease[J]. Cancer Res,2005,65(8):3100-3107. |
| [9] | Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction[J]. Anal Biochem, 1987, 162(1):156-159. |
| [10] | Schupp M, Kintscher U, Fielitz J, et al.Cardiac PPAR alpha expression in patients with dilated cardiomyopathy[J]. Eur J Heart Fail, 2006, 8(3):290-294. |
| [11] | Ye YM, Lin Y, Manickavasagam S, et al. Pioglitazone protects the myocardium against ischemia-reperfusion injury in eNOS and inos knockout mice[J]. Am J Physiol Heart Circ Physiol, 2008,295(6):H2436-H2446. |
| [12] | Ren L, Li Y, Li Y, et al. The inhibitory effects of rosiglitazone on cardiac hypertrophy through modulating the renin-angiotensin system in diet-induced hypercholesterolemic rats[J]. Cell Biochem Funct, 2001, 28(1):58-65. |
| [13] | Smeets PJH, Planavila A, Vusse GJ, et al. Peroxisome proliferator-activated receptors and inflammation: take it to heart[J]. Acta Physiol, 2007, 191(3):171-188. |
| [14] | Barcellos-Hoff MH. How do tissues respond to damage at the cellular level? The role of cytokines in irradiated tissues[J]. Radiat Res, 1998, 150(5):S109-120. |
| [15] | Wu R, Zeng Y. Does angiotensin II-aldosterone have a role in radiation-induced heart disease?[J] Med Hypotheses, 2009, 72(3):263-266. |
| [16] | Ghosh AK, Bhattacharyya S, Lakos G, et al. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma[J]. Arthritis Rheum, 2004, 50(4):1305-1318. |
| [17] | Guo B, Koya D, Isono M, et al. Peroxisome proliferator-activated receptor-Gamma ligands inhibit TGF-beta 1-induced fibronectin expression in glomerular mesangial cells[J]. Diabetes, 2004, 53(1): 200-208. |
| [18] | Chen K, Chen J, Li D, et al. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts: Modulation by PPARγ ligand pioglitazone[J]. Hypertension, 2004, 44 (5): 655- 661. |
| [19] | Wassmann S, Laufs U, Baumer AT, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species[J]. Hypertension, 2001, 37(6): 1450-1457. |
| [20] | Zhao SM, Shen LH, Li HW et al. Down regulation of the expression of angiotensin II type1 receptor in neonatal rat cardiac fibroblast by activation of PPAR gamma signal pathway[J]. Chin J Physiol, 2008, 51 (6): 357-362. |
| [21] | Hao GH, Niu XL, Gao DF, et al. Agonistsat PPAR gamma suppress angiotensin II induced production of plasminogen activator inhibitor 1 and extracel-lular matrix in rat cardiac fibroblasts[J]. Br J Pharmacol, 2008, 153 (7):1409-1419. |
| [22] | Gao S, Wu R, Zeng Y. Up-regulation of peroxisome proliferator-activated receptor gamma in radiation-induced heart injury in rats[J]. Radiat Environ Biophys, 2012,51(1):53-59. |
| [23] | Choi JH, Banks AS, Estall JL, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPAR gamma by Cdk5[J].Nature, 2010, 466(7305):451-457. |
| [24] | Dhavan R, Tsai LH. A decade of Cdk5[J]. Mol Cell Biol, 2001,2(10):749-759. |
| [25] | Camp HS, Whitton AL, Tafuri SR. PPAR-γ activators down-regulate the expression of PPAR-γ in 3T3-L1 adipocytes[J]. FEBS Lett,1999,447(2-3):186-190. |
| [26] | Benkirane K, Viel EC, Amiri F, et al. Peroxisome Proliferator-activated receptor-γ regulates angiotensin II-stimulated phosphatidylinositol 3-kinase and mitogen-activated protein kinase in blood vessels in vivo[J]. Hypertension, 2006, 47(1):102-108. |
| [27] | Chan SH, Wu KL, Kung PS, et al. Oral intake of rosiglitazone promotes a central antihypertensive effect via upregulation of peroxisome proliferator-activated receptor-γ and alleviation of oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats[J].Hypertension,2010, 55(6):1444-1453. |
| [28] | Alexis JD, Wang N, Che W, et al. Bcr kinase activation by angiotensin II inhibits peroxisome-proliferator -activated receptor transcriptional activity in vascular smooth muscle cells[J]. Circ Res,2009, 104(1):69-78. |
| [29] | Schoonjans K, Martin G, Staels B, et al. Peroxisome proliferator-activated receptors, orphans with ligands and functions[J]. Curr Opin Lipidol, 1997, 8(3):159-166. |
| [30] | Takeda K, Ichiki T, Tokunou T, et al. 15-Deoxy-delta 12,14-prostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phosphatidylinositol 3-kinase in vascular smooth muscle cells[J]. J Biol Chem, 2001, 276(52):48950-48955. |